5.3 MnClTPP self-assembly
Although the self-assembly of MnClTPP has been previously studied by Beggan
et al. [195], the following high-resolution images reveal some new behaviour not
reported by Beggan and colleagues.
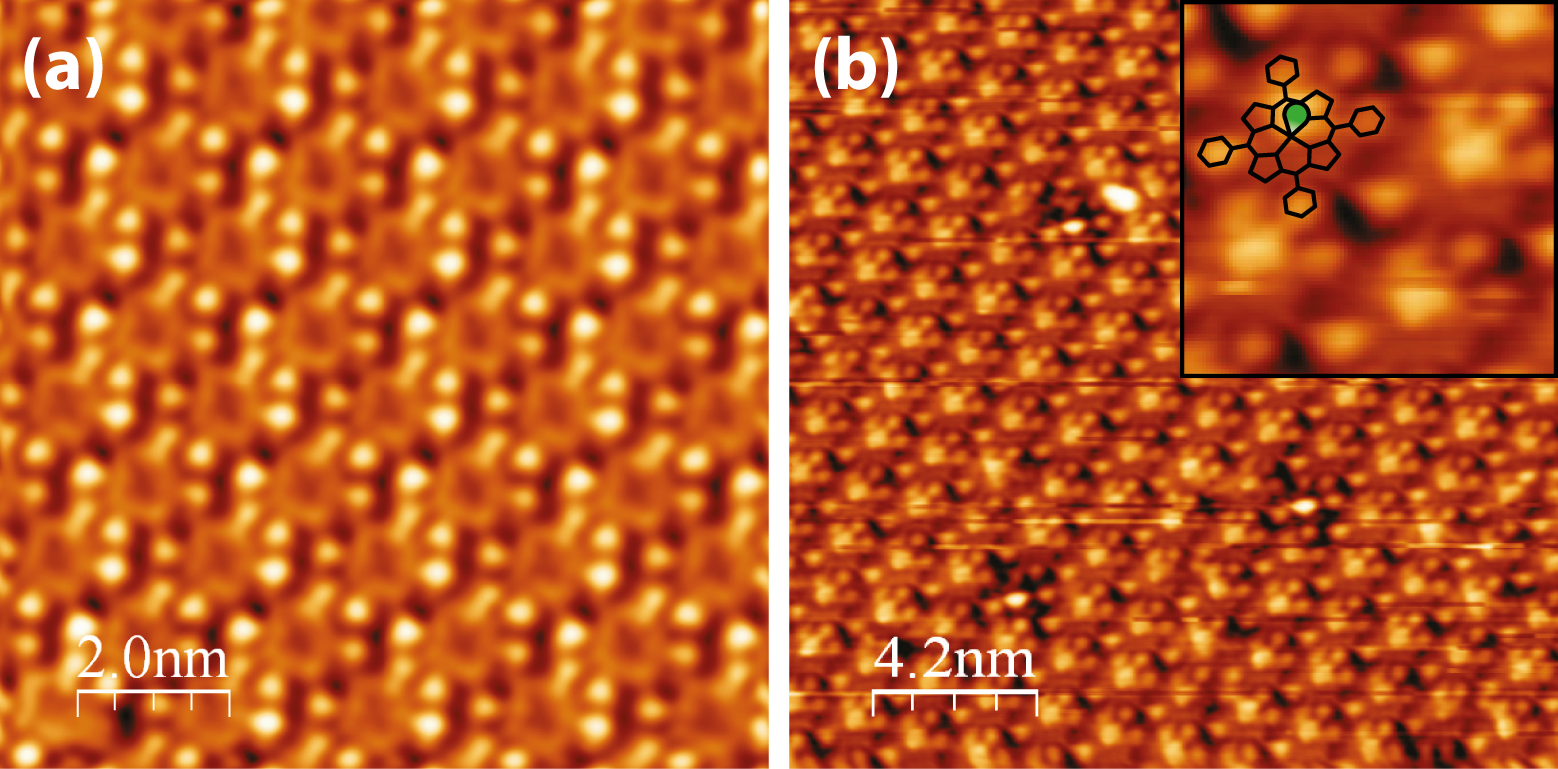
When deposited onto the Ag(111) surface, the MnClTPP molecules
self-assemble into close-packed structures (Figure 5.4). Although at low coverages
the Ag step edges are decorated, the majority of the growth occurs in the middle
of terraces, forming large, often rectangular islands, with a square packing
geometry and two equal lattice parameters of 1.41 ± 0.05 nm.
The supramolecular ordering of a deposited porphyrin overlayer has been
shown to be mostly triggered by the molecule’s side groups; indeed a similar
packing scheme, with the same geometry has previously been reported for
tetraphenyl-porphyrins (TPPs) with different metal centres on noble surfaces:
Co- and Fe-TPP on Ag(111) [196, 197]; and Ni-, Cu- and Co-TPP on
Au(111) [198], and the current data agree with those reported by Beggan et
al [195].
LEED taken from 1 ML of MnClTPP on the Ag(111) surface, published by
Beggan et al, shows a series of 12 spots arranged in a circle [195]. A similar LEED
pattern was observed for our system (not shown). These 12 spots are formed by 3
equivalent squares each rotated by 30°, corresponding to the three simple-cubic
domains of the MnClTPP monolayer.
One side of each square lies parallel to one of the close-packed directions of the
Ag(111) reciprocal unit cell, indicating that although the monolayer growth does
not begin at the step edges, in contrast to the case of NiDPP, the substrate still
plays a role in the self-assembly. Again, a similar arrangement has been observed
for other TPPs on Ag(111), with the substrate imparting a direction to the
overlayer primitive unit cell, and also one of the molecular axes lying parallel to a
close-packed Ag(111) direction [196, 197].
Although LEED indicates that three rotational domains coexist on
the surface, figure 5.4c shows the boundary between two domains in the
MnClTPP monolayer, with the same orientation of unit cells. It is evident
that the two domains are nevertheless different from one another in that
they cannot be transformed from one to the other by simple rotation or
translation, only by a mirror operation, i.e. they are chiral enantiomers of one
another.
It has previously been shown that achiral molecules deposited on an achiral
surface can give rise to chirality in the adsorbed layer [78, 216–218]. Indeed,
Buchner et al. have recently described the local organisational chirality of
TPP monolayers in some detail, and a similar treatment is applicable
here [194].
It is noted that in this study of MnClTPP, as in Buchner’s work, the only
domain boundaries observed within the closed monolayer were between chiral
domains with the same unit cell orientation. Boundaries between any of
the three orientational domains indicated by LEED were not observed
together on the same terrace, but monolayers with the three different
orientations were observed separately. This follows from Buchner’s hypothesis
that chiral domain boundaries, where the molecules are arranged in a
“zipper” fashion are more energetically favourable than orientational domain
boundaries, and that the activation energy for the rotation of an entire chiral
domain to align with its neighbouring domain is less than that required to
rotate all the individual molecules in a domain to give them the same
chirality.
The molecules are rotated by 15 ± 2° with respect to the close-packed
directions of the monolayer. Again, a similar azimuthal rotation has been observed
for Co- [194, 196], Fe- and free-base-TPP [194] on Ag(111), indicating
that this rotation arises from interactions between the TPP peripheral
groups.
Such a rotation allows the phenyl rings of adjacent molecules to interact in the
so-called “T-shape” configuration, where the edge of one phenyl ring is directed
toward the π-cloud on the face of its neighbouring phenyl ring [219]. This
accounts for a strongly attractive interaction between molecules and plays a major
role in their self-assembly, and has been noted to play a key role in both biological
and chemical recognition and the interactions between the aromatic side-chains of
proteins [220–222].
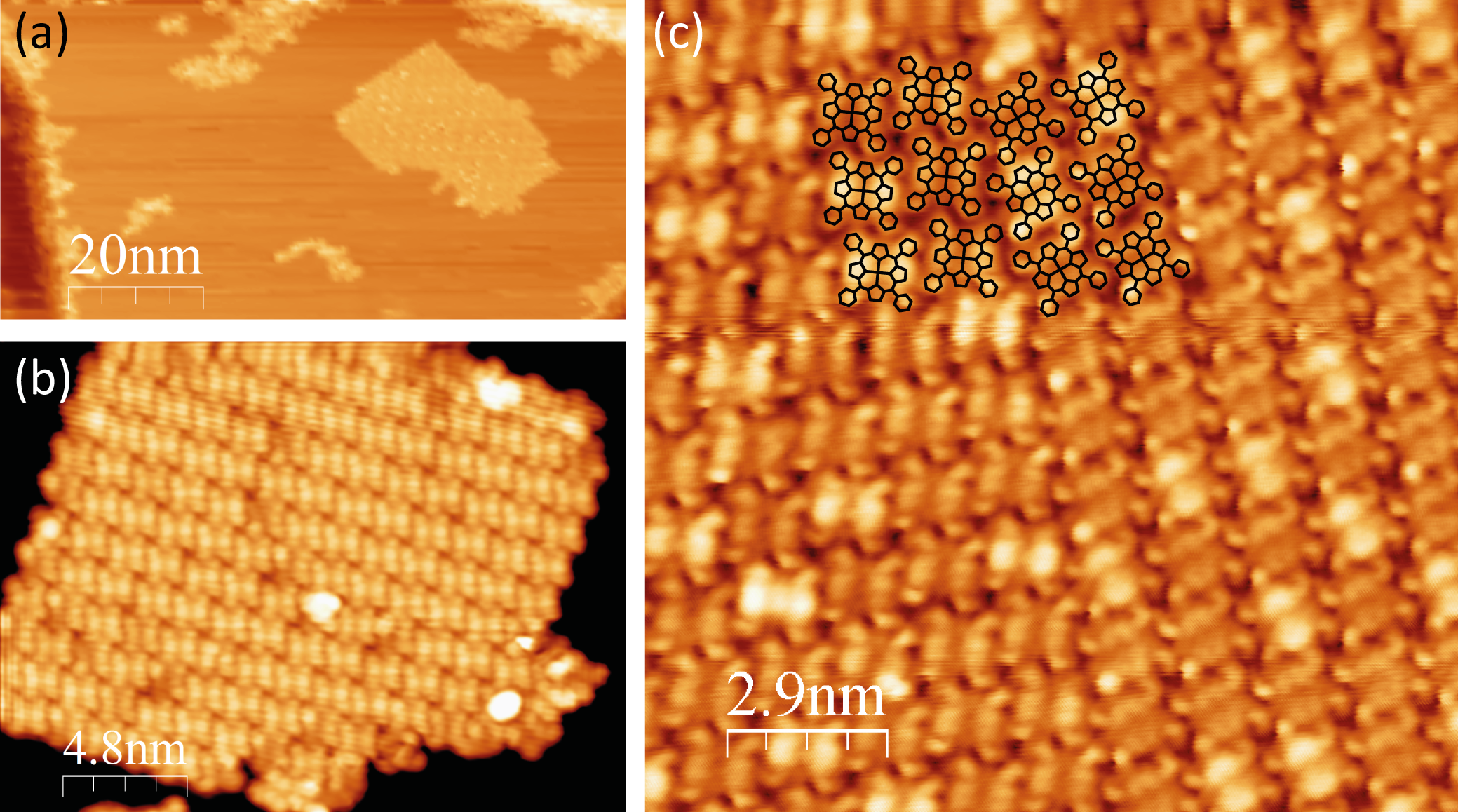
The molecules’ phenyl rings have an axis of rotation around the single C–C
bond joining them to the macrocycle. In order to match the molecular appearance
from STM images, two of the phenyl rings were rotated in the opposite direction
to the other two. We will refer to this as the “trans” conformation, shown in
Figure 5.5a (bottom), distinct from the “cis” conformation where all
four phenyl rings are rotated in the same direction, shown in Figure 5.5a
(top).
This “counter-rotation” gives rise to some steric hindrance due to the phenyl
rings’ proximity to the porphyrin centre. In turn, the macrocycle adopts a saddle
conformation, with the outer carbon and hydrogen atoms of the pyrrole rings
being pushed into or out of the plane of the molecule.
As an aside, one might naïvely assume that meso-aryl substituted porphyrins
in the gas phase would assume a conformation with the phenyl rings rotated by
90° to the porphyrin macrocycle in order to minimise steric hindrance. However,
steric hindrance is only half of the story. Calculated structures comparing H2P
(free-base porphine), H2TPP and their diacid analogues have shown that phenyl
substituent rotation is frequently accompanied by pyrrole ring saddling
in order to increase π-electron coupling between the phenyl rings and
the porphyrin macrocycle, reducing the overall potential energy of the
molecule [223].
Since the occupied state STM images of MnClTPP are dominated by the
phenyl rings, these features are used to support the saddling hypothesis. It is clear
from Figure 5.5b that the brightest (upper) portions of the phenyl rings are not
evenly spaced around the periphery of the molecule, as they would be in the “cis”
phenyl conformation. Instead, the bright protrusions are arranged at the
corners of a rectangle, implying that the upper parts of the phenyl rings are
tilted towards one another along the short side of the rectangle. This
conformation is supported by DFT, with the saddle-shaped molecule
(Figure 5.5a, bottom) having a total energy 5 meV/atom lower than the planar
conformation.
As shown in the schematic comparison between the upper STM image in
Figure 5.5b and the model below, this trans conformation reproduces the packing
of the molecules very well, with the upper-most parts of the phenyl rings
highlighted as these are the brightest features on the STM image due to their
proximity to the tip.
Such a saddling of the porphyrin molecule is generally accompanied by a
difference in the apparent height of the pyrrole rings’ features as observed by
STM [194], however at the sample bias Vsample = -1.4 V, the macrocycle appears
to have a mostly uniform height.
Also noted in STM images is some “buckling” in the monolayer. This is
illustrated in Figure 5.5c. The purple and green arrows point to two phenyl rings
with inequivalent heights within the same molecule.
This buckling of the monolayer can be described by a  ×
× R45° unit cell.
This is shown in black in Figure 5.5c, and the two inequivalent “types” of phenyl
rings are shown by purple and green circles.
R45° unit cell.
This is shown in black in Figure 5.5c, and the two inequivalent “types” of phenyl
rings are shown by purple and green circles.
A  ×
×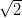 R45° unit cell was also suggested by Beggan and colleagues in
order for the molecular overlayer to lie commensurate to the Ag(111) surface, with
the corner molecules situated directly above a substrate atom and the central
molecule situated on a bridge site between two Ag atoms, however they did not
observe buckling in the monolayer [195].
R45° unit cell was also suggested by Beggan and colleagues in
order for the molecular overlayer to lie commensurate to the Ag(111) surface, with
the corner molecules situated directly above a substrate atom and the central
molecule situated on a bridge site between two Ag atoms, however they did not
observe buckling in the monolayer [195].
It is unclear whether this so-called “buckling” is caused by the plane of some
molecules deviating from a parallel orientation with the surface, or by one phenyl
ring of each molecule being tilted almost perpendicular to the molecular
plane, however from previous studies of porphyrin complexes on noble
metals [194, 196, 198], and the strength of the interaction between such
molecules and the surface, it is suggested that the latter is the case.
The interaction between the π-system of the macrocycle and the substrate has
already been mentioned, and its significance is seen in the fact that the molecular
unit cell and the axes of the molecules themselves are aligned along close-packed
directions of the surface. It is therefore clear that the strength of this interaction
is much larger than the energy required to rotate one phenyl ring around a single
C–C bond. This likely comes about in order to relieve some strain on the
molecular layer and to facilitate a closer edge–face interaction between
neighbouring phenyl rings.
5.3.1 Bias dependence of axial ligand resolution
Occupied-state STM images of the MnClTPP monolayer obtained at negative
sample biases (Figure 5.6a) show the molecules as four bright protrusions,
corresponding to the phenyl rings, surrounding the porphyrin core, which appears
as a darker ring with a dark centre. This is consistent with previous studies of
metallo-porphyrins showing that the occupied molecular orbitals are mostly
localised within the phenyl rings [195, 199–201]. Similarly, it has been shown for
Co-TBrPP that in the saddle conformation, the Co dz2 orbital did not contribute
to the HOMO of the molecule observed by STM [224], resulting in a
dark centre giving further evidence for the proposed MnClTPP saddle
conformation.
In contrast, when the unoccupied states of the molecules are probed at
positive a sample bias (Figure 5.6b), the molecules show a bright protrusion in
the centre corresponding to the Cl-ligand pointing out of the surface (green oval in
inset). In the previous study of this system, the Cl-ligand was not resolved,
however it is noted here that only under the specific tunneling conditions used in
Figure 5.6b was it possible to observe the axial ligand. Measurements were
performed at 78 K, and so the thermal stability of the molecules during the
current experiments was greater than that of Beggan and co-workers’ room
temperature work, also contributing to the ability to image the axial
ligand.
This is the first direct indication that the MnClTPP molecules arrive at the
Ag(111) surface with the Cl-ligand intact, however Beggan and co-authors have
shown that the chemical environment of the Cl atom is consistent with
coordination to the central Mn atom, and not reacted with the Ag(111)
surface [195]. They therefore propose that the Cl-ligand points out of the
surface into the vacuum, and the current results are in agreement with this
theory.
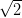 R45
R45