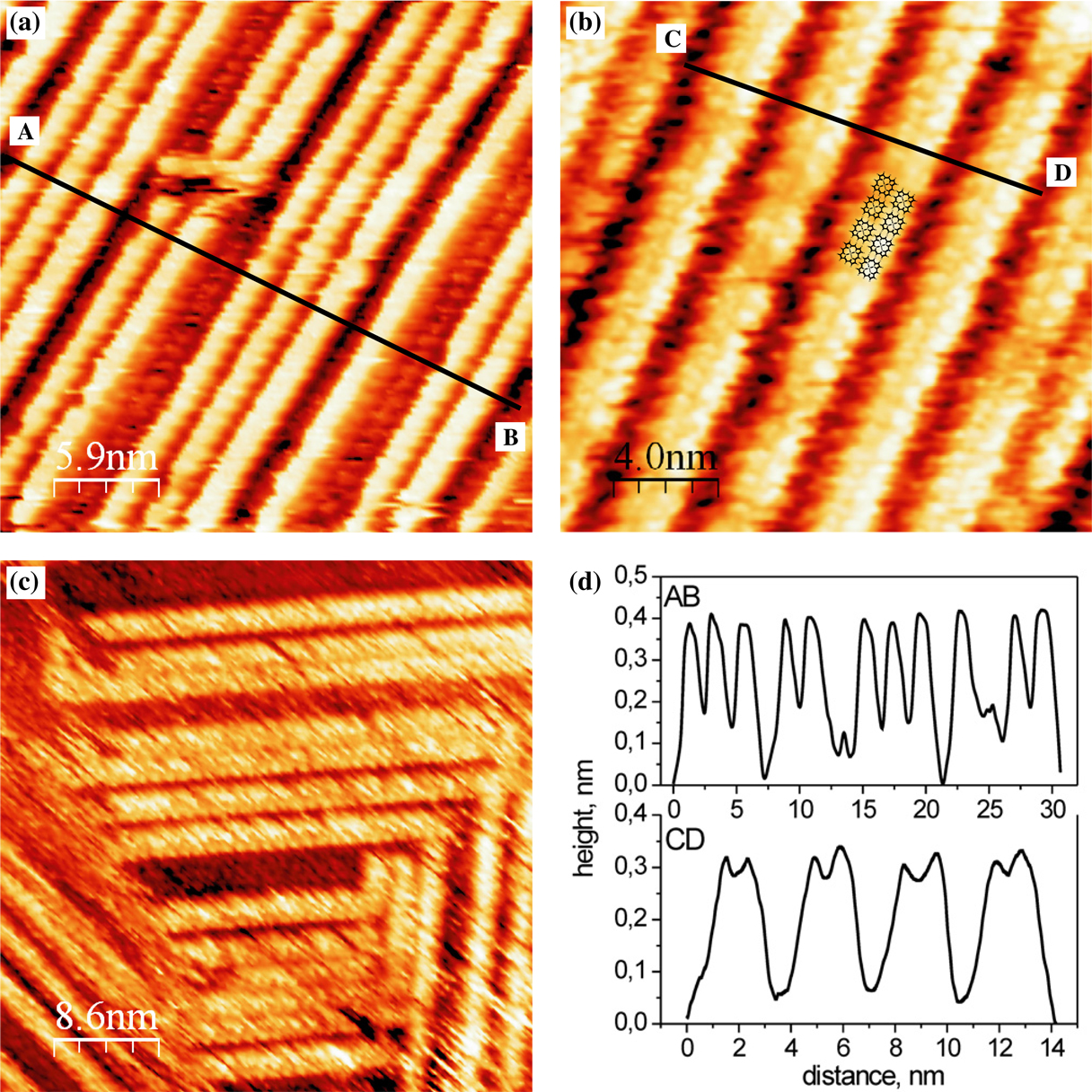
Figure 4.1: (a–c) STM of increasing NiP coverages on Ag(111), leading to
nanolines one (a), two (b) and three (c) molecules wide, as shown by the
line profiles in d [10].
Due to their interesting physicochemical properties and conformational flexibility, porphyrins are widely used for the fabrication of complex supramolecular structures, which are utilized in many technological applications including light-harvesting arrays for solar energy generation, sensors, molecular optoelectronic gates, photo-inducible energy or electron transfer systems, nonlinear optics and oxidation catalysts [1, 14, 78, 170–174]. Furthermore, they are essential to various natural biological processes as the main functional groups of haemoglobin and chlorophyll. To this end, porphyrin derivatives have been used to prepare a rich variety of molecular nanostructures such as clusters, wires and extended networks on different surfaces [1–13].
Such nanostructures offer a number of powerful approaches for the development of molecule-based devices [76–78]. The strength of the porphyrin-surface interaction is a vital parameter for the controlled assembly of these functional molecular species into ordered nanostructures [10, 80]. The nature of the bonding between porphyrins and the surface is reflected in the geometric configuration of molecules at the interface and their molecular charge distribution, and therefore can be probed by STM.
Since the porphyrin family comprises such a rich and diverse group of molecules, many moieties have been studied on many different substrates, from noble metals [8, 10, 11], to semiconductors [175, 176] and oxide surfaces [177, 178]. Several related classes of molecules are also well-studied on surfaces, such as phthalocyanine, porphyrazine, naphthalocyanine and anthrocyanine. Depending on the nature of the substrate and the structure of the porphyrin molecule, the supramolecular ordering can be vastly different, and the appearance of the molecules in STM is determined by the presence or absence of a metal centre.
On noble metal surfaces, such as Ag(111) and Au(111), molecule–molecule attraction tends to dominate over the relatively weak molecule–substrate interaction, and their self-assembly is largely determined by the side-groups present on the molecule.
Krasnikov et al. have shown the novel self-assembly of Ni-porphine (NiP) [10] and 5-((10,15,20-triphenylporphyrinato)Ni(II))2 dimer (NiTPP-dimer, structure shown in Figure 4.3a) [11] molecules on the Ag(111) surface.
Despite the fact that the two molecules investigated are both nickel porphyrins, even when deposited on a similar substrate they exhibit different self-assembly and packing, as defined by their individual structures.
It was shown that the structure of NiP layers depends very strongly on their coverage on the Ag(111) surface. At a coverage of 1 monolayer (ML), NiP forms a close-packed, uniform layer, with two equivalent domains rotated by ± 6° with respect to the Ag substrate. This is an example of non-chiral molecules self-assembling into right- and left-handed two-dimensional chiral domains.
At a coverage of approximately 1.5 ML, the second layer of NiP self-assembles into nanolines on top of the first molecular layer, as shown in Figure 4.1. These nanolines are very well-ordered and their length is limited only by the extent of the Ag substrate’s terraces, with lengths of up to 50 nm observed. They are found to be between one and four molecules wide, depending on the total molecular coverage, and as they grow wider they begin to form a hexagonal structure with unit cell parameters similar to those obtained for the first molecular layer.

As the coverage is further increased, the nanolines grow together laterally, and at 1.8 ML the molecules are seen to form lines with an average width of 5.0 ± 0.1 nm, with dark rows between them corresponding to a single missing molecular row (Figure 4.2). When the coverage finally reaches 2 ML, the closed hexagonal packing is restored, however the unit cell is approximately 4% larger than that of the first layer. This indicates that the molecule–substrate interaction experienced by the first layer is enough to accommodate this strain. Since it experiences less of an effect from the substrate, the second layer undergoes a lateral relaxation compared to the first layer, and this relaxation causes some of the molecules to be displaced out of plane, resulting in a 5.0 nm modulation of the apparent height, corresponding to the filling of the missing rows in going from 1.8 ML to 2.0 ML.
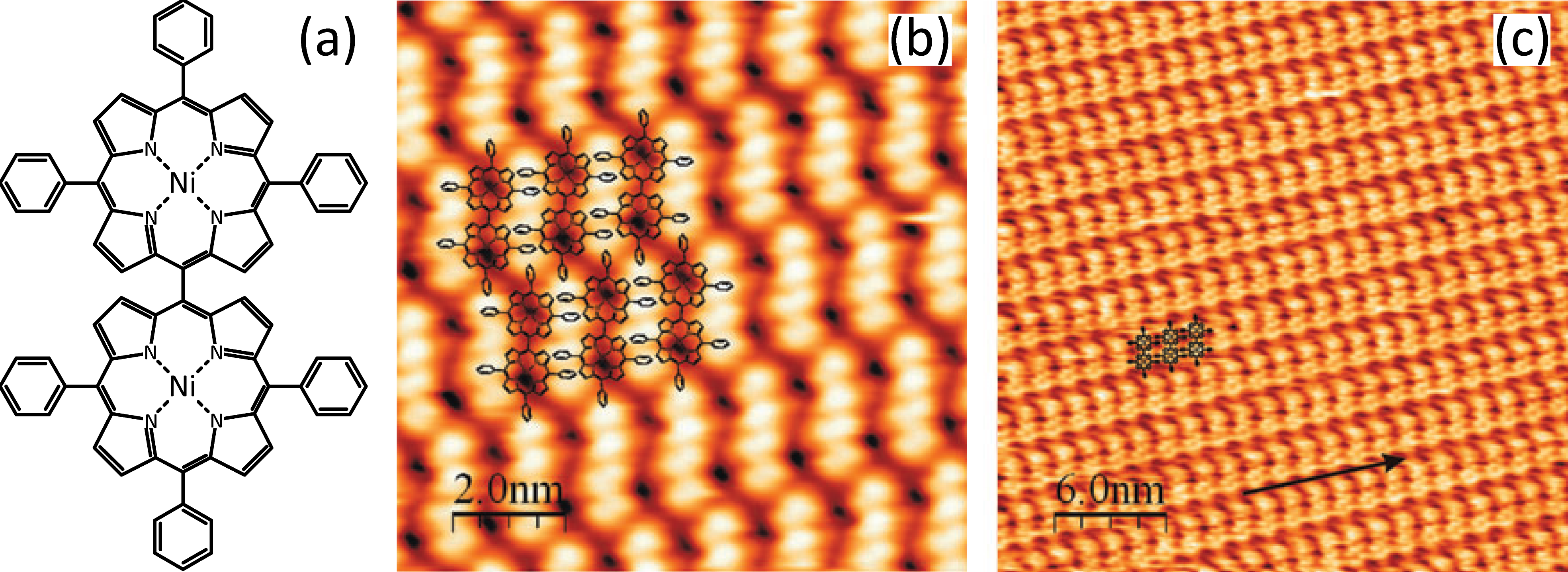
In the case of the NiTPP-dimer also deposited on the Ag(111) surface, however, a different behaviour has been reported [11]. The NiTPP-dimer consists of two nickel porphyrin macrocycles connected through a single carbon–carbon bond, with phenyl rings occupying the rest of the meso-positions, as shown in Figure 4.3a. This more complex structure gives rise to an oblique close packing regime, and at a coverage of 0.5 ML, large compact islands are formed, the edges of which appear “blurry” due to tip-induced molecular movement. This, and the relatively low desorption temperature of 500 K implies that they form a weakly bonded, physisorbed system.
When the coverage is increased to 1 ML the molecules form oblique closed domains, with lattice parameters of a = b = 2.7 nm and γ = 70°, and aligned along the main crystallographic directions of the Ag(111) surface. Figure 4.3b shows an STM image of such a monolayer, obtained at a bias voltage of Vsample = -1.9 V, where the central macrocycles appear as dark areas and the phenyl rings are imaged as bright protrusions. It can be seen that the dimers are very closely packed, with the phenyl rings rotated by 50–60°, allowing them to interlock and form intermolecular bonds through π-electron stacking of the phenyl rings.
By performing STM at a negative sample bias, as in Figure 4.3b, the occupied molecular orbital states are probed, and when tunneling at a positive sample bias, as in Figure 4.3c, the unoccupied states are examined. The published electronic structure of nickel porphyrin indicates that the first unoccupied molecular orbitals are localised within the macrocycle, and the occupied molecular orbitals within the phenyl substituents.
In contrast to noble metal surfaces, when deposited on bare or lightly-passivated semiconductor surfaces, organic molecular mobility is severely restricted even at room temperature, and isolated molecules can be stably imaged [179, 180]. This allows the conformational effects of the substrate to be probed without interactions from the molecule’s neighbours. On a highly boron-doped Si(111) surface, Cu-5,10,15,20-tetrakis(3,5-di- tert-butyl-phenyl) porphyrin (CuTBPP) strongly binds to the surface and undergoes some conformational adaptations due to the interaction with the substrate [180]. A similar pinning of the molecules is observed when phthalocyanines interact with defects on the H-passivated H:Si(001) surface [175, 176, 176].
On the passivated H:Si(001) and H:Si(111) surfaces, porphyrins and related organic molecules are free to migrate across the surface, similar to the noble metal surfaces, however when they encounter a defect they become strongly attached at the defect site [176, 181]. Voltage pulses from the STM tip can induce the formation of such defects by selectively removing H atoms, and in this way it has been hypothesised that nanoscale organic electronics can be designed [182, 183].
On oxidised surfaces such as Cu(110)-p(2 × 1)O, the inherently achiral free-base tetraphenyl porphyrin (H2TPP) forms heterochiral domains upon interaction with the surface. Although the H2TPP molecule is achiral, it consists of four phenyl rings which can be rotated in a “propeller” fashion with respect to the planar macrocycle either clockwise or anticlockwise, thus giving rise to chirality. The unit cell of the molecular monolayer on the oxide-reconstructed Cu surface consists of two molecules with opposite chirality. If the unit cell was square, this would lead to an overall-achiral monolayer, however on the Cu(110)-O surface, the commensurate unit cell is oblique, leading to the formation of mirror domains on adjacent terraces [78, 177].
As mentioned before, transition metal porphyrins possess many interesting properties, however in some cases their synthesis by traditional methods can be tricky, and some oxidation states are unstable in vitro. A solution to this is to directly metallate free-base porphyrin [184–186]. This versatile technique is employed most often by depositing a free-base porphyrin or phthalocyanine such as H2TPP onto the relatively-inert Ag(111) surface, and dosing the surface with metal atoms (e.g. Fe atoms to create Fe-TPP), and has shown yields of up to 95%, with almost all molecules converted to the metallated form [184, 185]. Conversely metal adatoms can be deposited on the surface before porphyrin deposition, and the metalation occurs in a similar way [186]. This novel on-surface synthesis can lead to the creation of organic molecules whose metal centres have oxidation states which are inaccessible to other synthesis methods, as they are stabilised through coordination by the surface.